Circos
circos 是一个关系可视化圈图表达软件,常被用来绘制环状的细菌染色体或质粒,或作为可视化比较基因组学研究多个细菌基因组之间差异的工具。circos 的优点是生成的图形优美,可以表示各种类型的数据。但是缺点在于牵涉内容过于繁杂,采用的术语名称的含义往往让人摸不着头脑。教程也不够明确。circos 的目的是绘制图形,有点类似Photoshop,它提供的是工具,而不是一步速成的获得某种图形的方法。在它的圈图体系中,各种 tracks 可以分成一下一种表现图示:
- Histogram 直方图
- Ideogram 染色体示意图
- inverted Histogram 反向直放图
- Heatmap 热力图
- Links 关系线
- Highlights 加亮图示
- Ticks 圈图标尺
- Grid 圈图网格
以mit的circos图为例,可以看出一个circos绘制的圈图中可以包含什么类型的图示。
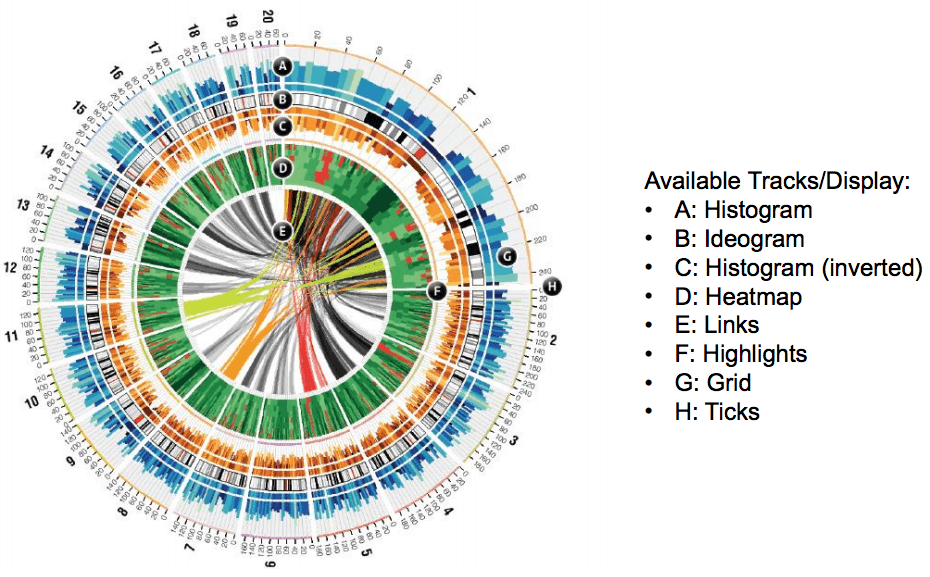
绘制一个circos图时,首先要考虑的是如何理解并展示复杂的数据关系。通过不同维度来解析数据,
$ circos -conf circos.conf
本节利用circos来简单演示几个可能会用到病原微生物成图功能。
1.安装 circos
1.1 安装依赖包
# install gd2-xpm development library
~$ sudo apt-get install libgd2-xpm-dev
# link circos perl import path. circos use "#!/usr/env perl" instead of "/usr/bin/env" which is default in ubuntu
~$ sudo ln -s /usr/bin/env /bin/env
# install perl modules needed in circos
~$ sudo cpan install Clone \
> Config::General \
> Font::TTF \
> List::MoreUtils \
> Math::Bezier \
> Math::VecStat \
> Math::Round \
> Params::Validate \
> Readonly \
> Regexp::Common \
> Set::IntSpan \
> Text::Format \
> GD \
> Statistics::Basic
1.2 安装circos
# download circos and extract
~$ wet http://circos.ca/distribution/circos-0.69.tgz
~$ tar xfz circos-0.69.tgz -C ~/apps/circos
# add circos to system
~$ sudo ln -s ~/apps/circos/bin/circos /usr/local/sbin/circos
# or you can add to your personal environment
~$ export PATH=$HOME/apps/circos/bin/
~$ circos -v
如果屏幕打印出以下信息,说明你可以正常使用circos了。
circos | v 0.69 | 6 Dec 2015 | Perl 5.018002
如果cpan下载很慢或者无法连接,可以修改mirror来改善。如果你的CPAN是安装在个人用户环境里,将~/.cpan/CPAN/MyConfig.pm文件里的urllist行后面的内容修改成离自己最近的镜像站点,比如:
'urllist' => [q[http://mirrors.sohu.com/CPAN/]],
2.配置文件
circos主要需要2类文件,一类是可视化结果的数据文件,一类是用于告诉circos如何展示的配置文件。
2.1 数据文件
由于circos只是画图工具,因此你图中要展示的数据需要自己预先准备好。比如基因组的GC含量,需要用其他程序先生成以下这种类型的文本格式文件。
染色体 开始位置 结束位置 颜色值 其他选项[可选]
chr1 1000 2000 red
2.2 配置文件
一般绘图需要以下配置文件:
- circos.conf
- ideogram.conf
- ticks.conf
- image.conf
- highlight.conf
- colors_fonts_patterns.conf
你可以将配置文件内容全部放在circos.conf里,也可以分离成单独的文件通过include方式导入。
2.2.1 circos.conf
这是circos主配置文件,其他的配置文件可以通过include的方式来导入。
<<include your_other_configure.conf>>
2.2.2 ideogram.conf
2.2.3 image.conf
2.2.4 highlight.conf
2.2.5 ticks.conf
2.3 可视化的组成要素
变量分为 Global 和 Local,对发挥作用的区块起作用。如果是某个变量在多个block中都重复使用,那么可以设置成 global 的参数,当某个block要使用新的值时,再用 local 的参数覆盖即可。
2.3.1 ideogram
2.3.2 highlights
2.3.3 ticks
2.3.4 label
2.3.5 links
2.3.6 circos.conf 参数
# 染色体的数据集
karyotype = data.txt
# 定义圈图标尺
chromosomes_units = 1000
# 如果设置成yes,所有ideogram上都会现实ticks
chromosomes_display_default = yes
2.3.7 完整circos配置文件结构
# ---- 定义图的颜色信息 ---- #
<colors>
</colors>
# ---- 定义图的字体信息 ---- #
<fonts>
</fonts>
# ---- 定义填充模式图 ---- #
<patterns>
</patterns>
# ---- 图像的基本设置信息 ---- #
<image>
</image>
# ---- ideogram 图 ---- #
<ideogram>
<spacing>
ideograms之间的间隔
<pairwise>
ideograms对(如人染色体)之间的距离
</pairwise>
</spacing>
</ideogram>
# ---- 标尺信息 ---- #
<ticks>
<tick>
各个tick的配置
</tick>
</ticks>
# ---- 区域的放大缩小信息 ---- #
<zooms>
<zoom>
</zoom>
</zooms>
# ---- highlights 数据 ---- #
<highlights>
<highlight>
<rules>
<rule>
某一个具体 data track 的配置修改
</rule>
</rules>
</highlight>
</highlights>
# ---- Data tracks 数据 ---- #
<plots>
<plot>
<rules>
<rule>
某一个具体 data track 的配置修改
</rule>
</rules>
<axes>
<axis>
plot 的半径轴
</axis>
</axes>
<backgrounds>
<background>
plot 的背景设置
</background>
</backgrounds>
</plot>
</plots>
# ---- links 数据 ---- #
<links>
<link>
<rules>
<rule>
某一个具体 link 数据的配置修改
</rule>
</rules>
</link>
</links>
2.3.8 circos配置的单位概念
一共有4种单位:p, r, u, b
- p表示像素,1p表示1像素
- r表示相对大小,0.95r表示95% ring 大小。
- u表示相对chromosomes_unit的长度,如果chromosomes_unit = 1000,则1u就是千分之一的染色体长度。
- b表示碱基,如果染色体长1M,那么1b就是百万分之一的长度。
3.用circos来绘图
3.1 绘制环状 Salmonella LT2 基因组
之前的教程里介绍了用DNAPlotter绘制一个基因组完成图的圈图,圆圈内容strand,GC content和GC skew。从某个角度上看,DNAplotter的配置文件与circos有一定的类似,2个软件的区别在于前者专注于绘制单个细菌基因组,后者可以完成更多的功能。第一个例子我们通过用circos来绘制类似DNAplotter生成的基因组圈图,从而学习circos的基本用法,
3.1.1 准备数据文件
从NCBI上下载Salonella LT基因组数据文件。
~$ esearch -db nuccore -query "NC_003197.1[accn]" | efetch -db nuccore -format fasta > LT2.fasta
~$ esearch -db nuccore -query "NC_003197.1[accn]" | efetch -db nuccore -format gb > LT2.gb
绘制circos所需要的ideogram所需要的几类数据,我们通过一个python脚本get\_circos\_data.py来获得。
# -*- utf-8 -*-
from Bio import SeqIO
rec = SeqIO.read("LT2.gb", "genbank")
forward = []
reverse = []
for feature in rec.features:
if feature.type != 'gene':
continue
if feature.strand == 1:
forward.append((int(feature.location.start), int(feature.location.end)))
elif feature.strand == -1:
reverse.append((int(feature.location.start), int(feature.location.end)))
def get_forward_strand(strands):
"""
Create forward strands
"""
with open("forward.txt", "w") as f:
for strand in strands:
f.writelines("chr1\t%d\t%d\tfillcolor=chr4\n" % (strand[0], strand[1]))
def get_reverse_strand(strands):
"""
Create forward strands
"""
with open("reverse.txt", "w") as f:
for strand in strands:
f.writelines("chr1\t%d\t%d\tfillcolor=chr6\n" % (strand[0], strand[1]))
def calc_GC_content(seq):
"""
Return GC content of input sequence
"""
gc = sum(seq.count(x) for x in ['G','C','g','c'])
gc_content = gc/float(len(seq))
return round(gc_content, 4)
def calc_GC_skew(seq):
"""
Reuturn GC skew of input sequence
"""
g = seq.count('G') + seq.count('g')
c = seq.count('C') + seq.count('c')
try:
skew = (g - c)/float(g + c)
except ZeroDivisionError:
skew = 0
return round(skew, 4)
window = 10000
step = 5000
seqs = SeqIO.parse("LT2.fasta", "fasta")
for seq in seqs:
seq_1 = seq.seq
with open("gc_skew.txt", "w") as f1:
with open("gc_content.txt", "w") as f2:
for i in range(0, len(seq_1), step):
subseq = seq_1[i:i+window]
gc_content = (calc_GC_content(subseq))
gc_skew = (calc_GC_skew(subseq))
start = (i + 1 if (i+1<=len(seq_1)) else i)
end = ( i + step if (i+ step<=len(seq_1)) else len(seq_1))
f1.writelines("chr1\t%d\t%d\t%s\n" % (start, end, gc_skew))
f2.writelines("chr1\t%d\t%d\t%s\n" % (start, end, gc_content))
get_forward_strand(forward)
get_reverse_strand(reverse)
运行脚本:
~$ python get_circos_data.py
3.3.2 配置文件
建立circos.conf配置文件
~$ mkdir circos_data && cd circos_data
~/circos_data$ touch circos.conf
大部分细菌的基因组都是单染色体的,因此一个细菌基因完成图可以用1个完整的圈来表示。通过设置spacing来去除间断。
<spacing>
default = 0u
break = 0u
</spacing>
circos.conf 文件添加以下内容
<<include colors_fonts_patterns.conf>>
<ideogram>
<spacing>
default = 0u
break = 0u
</spacing>
radius = 0.85r
thickness = 5p
stroke_thickness = 1
stroke_color = black
fill = yes
fill_color = black
</ideogram>
<image>
dir = .
file = circos.png
png = yes
svg = yes
# radius of inscribed circle in image
radius = 1000p
# by default angle=0 is at 3 o'clock position
angle_offset = -90
#angle_orientation = counterclockwise
auto_alpha_colors = yes
auto_alpha_steps = 5
background = white
</image>
karyotype = data/node.txt
chromosome_units = 1000
chromosomes_display_default = yes
show_ticks = yes
show_tick_labels = yes
show_grid = no
grid_start = dims(ideogram,radius_inner) - 0.5r
grid_end = dims(ideogram,radius_inner)
# --------------------- ticks start --------------------- #
<ticks>
# if yes, skip of label of 0bp
skip_first_label = no
# if yes, skip of lable of last
skip_last_label = no
# ticks radius to show, outside or inside
radius = dims(ideogram,radius_outer)
# genome size * label_multiplier = label units
label_multiplier = 1e-6
tick_separation = 2p
min_label_distance_to_edge = 0p
label_separation = 5p
# label distance to ticks
label_offset = 5p
label_size = 8p
size = 20p
<tick>
spacing = 10000u
color = gray
show_label = no
thickness = 1p
</tick>
<tick>
spacing = 100000u
color = black
show_label = yes
label_size = 16p
thickness = 2p
# label format, %.1f = 1.1, %d = 1, %.2f = 1.10
format = %.1f
grid = yes
grid_color = white
grid_thickness = 1p
</tick>
<tick>
spacing = 1000000u
size = 30p
color = black
show_label = yes
label_size = 24p
thickness = 3p
# label format, %.1f = 1.1, %d = 1, %.2f = 1.10
format = %d
# plus Mb to lables
suffix = " Mb"
grid = yes
grid_color = dgrey
grid_thickness = 1p
</tick>
</ticks>
# --------------------- ticks ends ---------------------- #
# --------------------- plot start ---------------------- #
<plots>
<plot>
type = tile
file = data/forward.txt
r1 = 0.95r
r0 = 0.90r
margin = 1b
stroke_thickness = 0
color = orange
orientation = center
# tile height
thickness = 20p
# different layer index height
padding = 5p
layers = 1
<rules>
<rule>
condition = var(size) > 5kb
color = black
</rule>
</rules>
</plot>
<plot>
type = tile
file = data/reverse.txt
r1 = 0.92r
r0 = 0.87r
margin = 1b
stroke_thickness = 0
color = blue
thickness = 20p
padding = 5p
# display one layer of plot
layers = 1
<rule>
condition = var(size) > 3kb
color = black
</rule>
</plot>
<plot>
type = line
r1 = 0.85r
r0 = 0.75r
file = data/gc_skew.txt
thickness = 1
max_gap = 1u
color = vdgrey
# max/min value of line plotting
min = -0.15
max = 0.15
fill_color = vdgrey_a3
<backgrounds>
<background>
color = vvlgreen
y0 = 0.05
</background>
<background>
color = vvlred
y1 = -0.05
</background>
</backgrounds>
<axes>
<axis>
color = lgrey_a2
thickness = 1
spacing = 0.04r
</axis>
</axes>
</plot>
<plot>
type = histogram
r1 = 0.70r
r0 = 0.60r
file = data/gc_content.txt
thickness = 1
max_gap = 1u
min = 0
max = 1
<rules>
<rule>
condition = 1
fill_color = eval(sprintf("spectral-9-div-%d", remap_int(var(value), 0,1,1,9)))
</rule>
</rules>
</plot>
</plots>
<<include housekeeping.conf>>
3.2 绘制测序拼接结果信息
3.2.1 数据准备
3.2.2 配置文件
3.3. 绘制一个比较基因组学数据
BRIG 常被用来绘制的多个细菌比较基因组学数据。本例用circos绘制类似的圈图,通过结果可视化来发现和展示生物学信息。